|
Our company offers unique receptor-based targeted libraries. They are produced based on computational estimation of compounds’ interaction with one specific member in a protein family (sharpfocusing approach). Each library contains about 2,000 compounds selected from OTAVAchemicals collection of 260,000 in-stock compounds.
Read more: List of molecular targets suitable for receptor-based virtual screening
WHY OTAVAchemicals TARGETED LIBRARIES?
Our proprietary method for receptor-based virtual screening of compounds includes powerful combination of drug-likeness filtering, molecular docking, re-scoring, key intermolecular hydrogen bond detection and,finally, visual inspection of ligand-receptor complexes. The main advantage of our targeted libraries is that each compound in every library is finally selected manually:
-
using drug-likeness filtering, molecular docking, re-scoring and H-bond detection scientists initially reduce input library of in-stock compounds to about 10,000 and then
-
a panel of our experts analyzes these compounds manually. They visually inspect about 10,000 ligand-receptor complexes for proper binding mode: hydrogen bonding, hydrophobic interactions as well as orientation of ligands in a binding site, and finally
-
look into the diversity of chemical scaffolds
It is demanding, but in this way we could insure that our customers will get the most promising candidate molecules for their specific tasks. Our approach works and our scientists already found a number of new kinase inhibitors with high activity and selectivity [1-4].
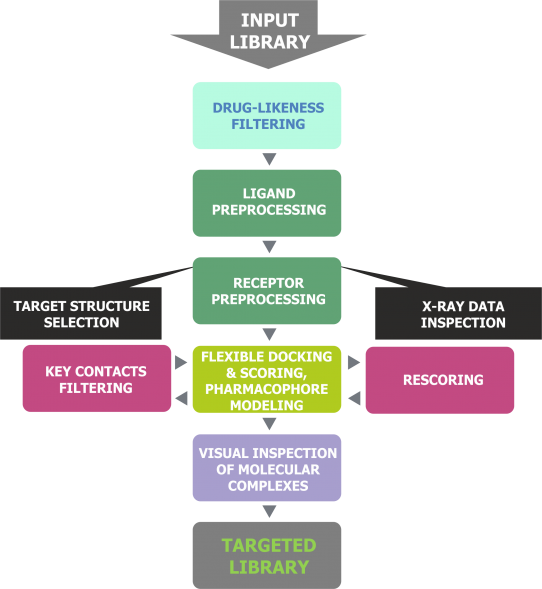
Fig. 1. Design flowchart of receptor-based targeted libraries.
OTAVAchemicals UNIQUE CALCULATION ALGORITHM
To prepare these targeted libraries, our experts have implemented a number of crucial improvements of a calculation algorithm which include calculation of atom charges as well as unique force field and scoring functions [5-7]. Taking into account key electrostatic interactions between molecules, we developed a specific chargedefinition algorithm which uses Kirchhoff method for electronegativity relaxation. This allowed us to treat electrostatic terms more accurately. To achieve reasonable acceleration in entropy loss measurement, we applied harmonic approximation for integration of configuration space of a ligand-receptor complex. Scoring function also requires co-crystallized water environment to be efficient. Therefore, we developed new method to calculate and predict binding of water molecules to the formed complex on sub-second timescale.
MOLECULAR DYNAMICS
As part of our Contract Research projects we also utilize MD simulation for limited number of complexes to study their binding modes in more detail. This approach allows us to make right decisions on further structural optimization of lead molecules.
REFERENCES (PUBLISHED BY OTAVAchemicals SCIENTISTS)
-
Volynets G, Vyshniakova H, Nitulescu G, Nitulescu, GM, Ungurianu A, Margina D, Moshynets O, Bdzhola V, Koleiev I, Iungin O, Tarnavskiy S, Yarmoluk S (2021) Identification of Novel Antistaphylococcal Hit Compounds Targeting Sortase A. Molecules 26: 7095.
-
Rybak MY, Balanda AO, Yatsyshyna AP, Kotey IM, Starosyla SA, Bdzhola VG, Lukash LL, Yarmoluk SM, Tukalo MA, Volynets GP (2021) Discovery of novel antituberculosis agents among 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives targeting aminoacyl-tRNA synthetases. Scientific Reports, 11(1), 7162.
-
Volynets GP, Vdovin VS, Lukashov SS, Oleksiy Borovykov, Borysenko IP, Gryshchenko AA, Bdzhola VG, Iatsyshyna AP, Lukash LL, Bilokin YV, Yarmoluk SM (2021) Identification of Novel Indazole-based Inhibitors of Fibroblast Growth Factor Receptor 1 (FGFR1). Current Enzyme Inhibition.
-
Protopopov MV, Vdovin VS, Starosyla SA, Borysenko IP, Prykhod'ko AO, Lukashov SS, Bilokin YV, Bdzhola VG, Yarmoluk SM (2020) Flavone inspired discovery of benzylidenebenzofuran-3(2H)-ones (aurones) as potent inhibitors of human protein kinase CK2. Bioorg Chem 102:104062.
-
Kovalenko OP, Volynets GP, Rybak MYu, Starosyla SA, Gudzera OI, Lukashov SS, Bdzhola VG, Yarmoluk SM, Boshoff HI, Tukalo MA (2019) Dual-target inhibitors of mycobacterial aminoacyl-tRNA synthetases among N-benzylidene-N'-thiazol-2-yl-hydrazines. MedChemComm 10:2161-2169.
-
Volynets GP, Lukashov SS, Borysenko IP, Gryshchenko AA, Starosyla SA, Bdzhola VG, Ruban TP, Iatsyshyna AP, Lukash LL, Bilokin YV, Yarmoluk SM (2019) Identification of protein kinase fibroblast growth factor receptor 1 (FGFR1) inhibitors among the derivatives of 5-(5,6-dimethoxybenzimidazol-1-yl)-3-hydroxythiophene-2-carboxylic acid. Monatshefte fur Chemie - Chemical Monthly 150:1801-1808.
-
Kotey IM, Protopopov MV, Starosyla SA, Balanda AO, Pletnova LV, Prykhod’ko AO, Bdzhola VG, Yarmoluk SM (2019) Identifiation of 4-methoxythieno[2,3-d]pyrimidines as FGFR1 Inhibitors. Biopolym Cell 35(2):152-162.
-
Tarnavskiy SS, Protopopov MV, Borovykov OV, Pryhodko AO, Bdzhola VG, Yarmoluk SM (2019) Hit identification of FGFR1 inhibitors using receptor-based virtual screening. Biopolym Cell 35(2):143-151.
-
Protopopov MV, Volynets GP, Starosyla SA, Vdovin VS, Lukashov SS, Bilokin YV, Bdzhola VG, Yarmoluk SM (2018) Identification of 1,3-thiazole-5-carboxylic acid derivatives as inhibitors of protein kinase CK2. Curr Enzyme Inhib 14(2):152-159.
-
Protopopov MV, Ostrynska OV, Starosyla SA, Vodolazhenko MA, Sirko SM, Gorobets NY, Bdzola VG, Desenko SM, Yarmoluk SM (2018) Dihydrobenzo[4,5]imidazo[1,2-a]pyrimidine-4-ones as a new class of CK2 inhibitors. Mol Div Nov;22(4):991-998.
-
Niefind K, Bischoff N, Golub AG, Bdzhola VG, Balanda AO, Prykhod'ko AO, Yarmoluk SM (2017) Structural hypervariability of the two human protein kinase CK2 catalytic subunit paralogs revealed by complex structures with a flavonol- and a thieno[2,3-d]pyrimidine-based inhibitor. Pharmaceuticals (Basel) 10(1):pii:E9.
-
Zinchenko AN, Muzychka LV, Smolii OB, Bdzhola VG, Protopopov MV, Yarmoluk SM (2017) Synthesis and biological evaluation of novel amino-substituted derivatives of pyrido[2,3-d]pyrimidine as inhibitors of protein kinase CK2. Biopolym Cell 33(5):367-378.
-
Syniugin AR, Ostrynska OV, Chekanov MO, Volynets GP, Starosyla SA, Bdzhola VG, Yarmoluk SM (2016) Design, synthesis and evaluation of 3-quinoline carboxylic acids as new inhibitors of protein kinase CK2. J Enzyme Inhib Med Chem 31(sup4):160-169.
-
Gudzera OI, Golub AG, Bdzhola VG, Volynets GP, Kovalenko OP, Boyarshin KS, Yaremchuk AD, Protopopov MV, Yarmoluk SM, Tukalo MA (2016) Identification of Mycobacterium tuberculosis leucyl-tRNA synthetase (LeuRS) inhibitors among the derivatives of 5-phenylamino-2H-[1,2,4]triazin-3-one. J Enzyme Inhib Med Chem 31(sup2):201-207
-
Ostrynska OV, Balanda AO, Bdzhola VG, Golub AG, Kotey IM, Kukharenko OP, Gryshchenko AA, Briukhovetska NV, Yarmoluk SM (2016) Design and synthesis of novel protein kinase CK2 inhibitors on the base of 4-aminothieno[2,3-d]pyrimidines. Eur J Med Chem 115:148-160.
-
Gryshchenko AA, Tarnavskiy SS, Levchenko KV, Bdzhola VG, Volynets GP, Golub AG, Ruban TP, Vygranenko KV, Lukash LL, Yarmoluk SM. Design, synthesis and biological evaluation of 5-amino-4-(1H-benzoimidazol-2-yl)-phenyl-1,2-dihydro-pyrrol-3-ones as inhibitors of protein kinase FGFR1. Bioorg Med Chem 2016, 24 (9), 2053-2059.
-
Gudzera OI, Golub AG, Bdzhola VG, Volynets GP, Lukashov SS, Kovalenko OP, Kriklivyi IA, Yaremchuk AD, Starosyla SA, Yarmoluk SM, Tukalo MA. Discovery of potent anti-tuberculosis agents targeting leucyl-tRNA synthetase. Bioorg Med Chem, 2016, 24 (5), 1023-1031.
-
Starosyla SA, Volynets GP, Lukashov SS, Gorbatiuk OB, Golub AG, Bdzhola VG, Yarmoluk SM. Identification of apoptosis signal-regulating kinase 1 (ASK1) inhibitors among the derivatives of benzothiazol-2-yl-3-hydroxy-5-phenyl-1,5-dihydro-pyrrol-2-one. Bioorg Med Chem, 2015, 23 (10), 2489-2497.
-
Guerra B, Bischoff N, Bdzhola VG, Yarmoluk SM, Issinger OG, Golub AG, Niefind K. A note of caution on the role of halogen bonds for protein kinase/inhibitor recognition suggested by high- and low-salt CK2α complex structures. ACS Chem Biol, 2015, 10 (7), 1654-1660.
-
Hunnigan K, Kulkarni SS, Bdzhola VG, Golub AG, Yarmoluk SM, Talele TT. Identification of novel PARP-1 inhibitors by structure-based virtual screening. Bioorg Med Chem Lett, 2013, 23, 5790-5794.
-
Golub AG, Gurukumar KR, Basu A, Bdzhola VG, Bilokin Y, Yarmoluk SM, Lee J-C, Talele TT, Nichols DB, Kaushik-Basu N. Discovery of New Scaffolds for Rational Design of HCV NS5B Polymerase Inhibitors. Eur J Med Chem, 2012, 58, 258-264.
-
Sapelkin et al. Application of 4-substituted 3-carboxyquinolines as protein kinase CK2 inhibitors. Pat. UА68984 А, C07D215/00, 2004-08-16.
-
Prykhod'ko et al. Application of 4,5,6,7-tetrahalogeno-1,3-isoindolinediones as protein kinase CK2 inhibitors. Pat. UA69165 А, С07D215/00, 2004-08-16.
-
Yakovenko et al. Kirchhoff atomic charges fitted to multipole moments: implementation for a virtual screening system. Journal of Computational Chemistry, 2008, 29 (8), 1332-1343.
-
Yakovenko et al. The new method of distribution integrals evaluations for high throughput virtual screening. Ukrainica Bioorganica Acta, 2007, 5 (1), 52-62.
|
 HOME
HOME Overview
Overview