|
2-({2-[(3-fluorophenyl)amino]pyrimidin-4-yl}amino)benzoic acid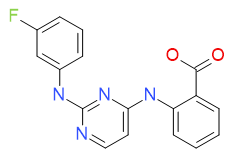
JNK inhibitor (IC50 = 28 nM)
Chemical Formula: C17H13FN4O2
Molecular Weight: 324.3172
OTAVAchemicals Catalogue Number: 6543537
CAS Registry Number: 929007-58-9
Purity: 95% (HPLC)
Ref. 1. Liu, M., Wang, S., Clampit, J. E., Gum, R. J., Haasch, D. L., Rondinone, C. M., … Liu, G. Discovery of a new class of 4-anilinopyrimidines as potent c-Jun N-terminal kinase inhibitors: Synthesis and SAR studies. Bioorganic & Medicinal Chemistry Letters (2007), 17(3), 668–672.
Ref. 2. Simon-Szabó, L., Kokas, M., Greff, Z., Boros, S., Bánhegyi, P., Zsákai, L., … Kéri, G. Novel compounds reducing IRS-1 serine phosphorylation for treatment of diabetes. Bioorganic & Medicinal Chemistry Letters (2016), 26(2), 424–428.
Ref. 3. T. Madhavan, J. Y. Chung et al. 3D-QSAR Studies of JNK1 Inhibitors Utilizing Various Alignment Methods. Chem Biol Drug (2012), 79, 53-67.
Abstract 1. A new series of 4-anilinopyrimidines has been synthesized and evaluated as JNK1 inhibitors. SAR studies led to the discovery of potent JNK1 inhibitors with good enzymatic activity as well as cellular potency represented by compound 2b. Kinase selectivity profile and the crystal structure of 2b are also described.
Abstract 2. Activation of various interacting stress kinases, particularly the c-Jun N-terminal kinases (JNK), and a concomitant phosphorylation of insulin receptor substrate 1 (IRS-1) at serine 307 play a central role both in insulin resistance and in ß-cell dysfunction. IRS-1 phosphorylation is stimulated by elevated free fatty acid levels through different pathways in obesity. A series of novel pyrido[2,3-d]pyrimidin-7-one derivatives were synthesized as potential antidiabetic agents, preventing IRS-1 phosphorylation at serine 307 in a cellular model of lipotoxicity and type 2 diabetes.
Abstract 3. We report our three-dimensional quantitative structure activity relationship (3D-QSAR) studies of the series of anilinopyrimidine derivatives of JNK1 inhibitors. The comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were applied using different alignment methods. The ligand-based atom-byatom matching alignment has produced better values for CoMFA (q2 = 0.646 and r2 = 0.983), while in CoMSIA it has achieved only lower statistical values. The pharmacophore-based model has produced (q2 = 0.568, r2 = 0.938) and (q2 = 0.670, r2 = 0.982) for CoMFA and CoMSIA models, respectively. As the model was based on the receptor-guided alignment, all the compounds were optimized within the receptor, resulting in q2 = 0.605 and r2 = 0.944 for CoMFA, and q2 = 0.587 and r2 = 0.863 for CoMSIA. Molecular Dynamic simulation studies suggested that the generated models were consistent with the low-energy protein ligand conformation. The CoMFA and CoMSIA contour maps indicated that the substitutions of the electropositive groups in the phenyl ring, and an addition of hydrophobic groups in the pyrimidine ring, are important to enhance the activity of this series. Moreover, the virtual screening analysis against NCI database yields potentials hits, and the results obtained would be useful to synthesize selective and highly potent c-Jun N-terminal kinase 1 analogs.
DOI:
10.1016/j.bmcl.2006.10.093
10.1016/j.bmcl.2015.11.099
10.1111/j.1747-0285.2011.01168.x
Price info:
|
1 MG |
49 EUR |
|
5 MG |
79 EUR |
|
10 MG |
109 EUR |
|
300uL of 10mM solution |
64 EUR
|
|
 HOME
HOME Overview
Overview